

How to use BEscreen
We're happy you found your way to BEscreen. BEscreen lets you easily design base editing libraries for screens of pre-specified variants. It can also generate libraries to introduce any possible edit in a given gene, transcript or genomic region (saturation screens). With BEscreen, you can also design guides that introduce an edit with a given consequence, such as guides that lead to missense, non-sense, splice-site edits etc. For example, this way, you can use BEscreen to design negative controls (non-editing or synonymously editing guides) for new and already existing libraries.
Of course you can also design individual guides for single edits. BEscreen accepts several input formats: if you want to screen for given variants ("variant mode"), you can input genomic variants, protein amino acid changes, and rsIDs from dbSNP. If you want to generate guides for a given genomic region (e.g., all coding sequences of a given gene; "gene/region mode"), you input gene names, transcript names or genomic regions.
Finally, BEscreen allows for comprehensive customization of your editing tools - select one of the pre-specified presets to set base editor options or input the characteristics of your base editor of choice (including customizing the editing window, fully customizable PAM site etc.).
To get started with BEscreen, follow the flow diagram to find out which mode you need to use and start designing your base editing library!

Entering your data and options
This is how it looks:
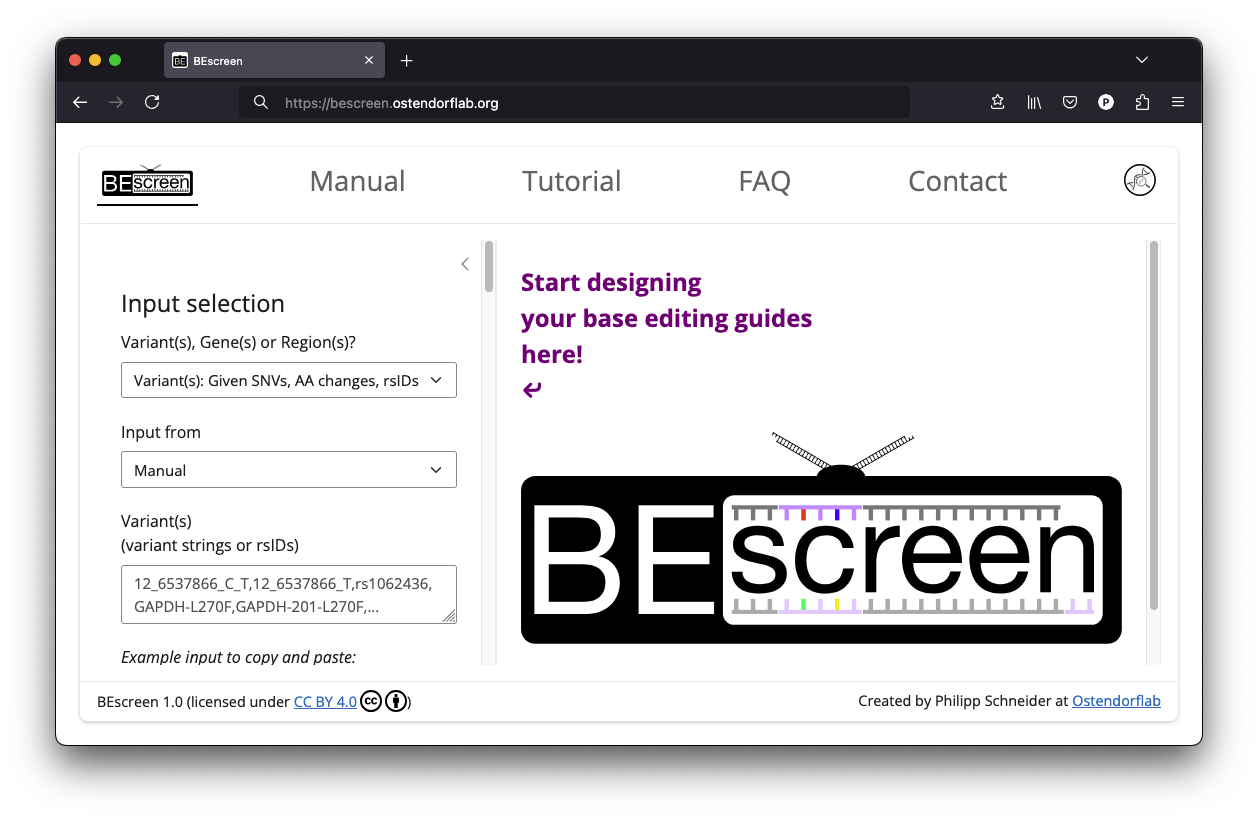
Input selection
Variant(s)
If you want to perform a screen for a list of specific variants, select Variant(s): Given SNVs, AA changes, rsIDs as input. As the option indicates, BEscreen allows you to enter your variants as genomic variants (e.g., 12_6537866_C_T), as protein amino acid changes using gene or transcript names (e.g., GAPDH-L270F or GAPDH-201-L270F), or as an rsID (e.g., rs1062436) as outlined in more detail below. You can mix different input formats.
You can choose whether you want to directly enter the given variants in the corresponding field like this:

or to upload a CSV file containg your variants of interest:
Direct variant inputs need to be provided either as:
-
genomic positions:
[chromosome]_[genomic_position]_[alternative_base] or [chromosome]_[genomic_position]_[reference_base]_[alternative_base]
e.g.: 12_6537866_C_T or 12_6537866_T -
rsID:
e.g.: rs1062436 -
protein amino acid changes
[gene_symbol]-[transcript_number]-[WTAApositionMUTAA] or [gene_symbol]-[transcript_number]-[WTAApositionMUTAA]
e.g.: GAPDH-L270F or GAPDH-201-L270F
If you don't provide a transcript number the MANE select transcript will be used.
e.g. like this:

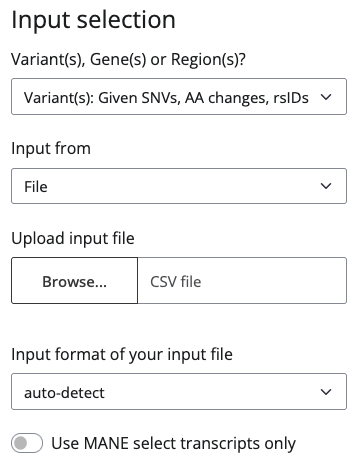
If you choose to input from file, you can use the columns variant or chr, pos, ref and alt. BEscreen will detect your columns automatically if you only provide one option. If your file contains all five columns, you need to specify which columns to use in the Input format field. The column variant can be populated by genomic positions (joined by underscores, rsIDs or protein amino acid changes). The columns chr, pos, ref and alt can only be populated by individual genomic positions.
Thus, CSV files containing variants need to be formatted as either of the following examples:
-
columns
variantvariant 12_6537866_C_T 12_6537866_T rs1062436 GAPDH-L270F GAPDH-201-L270F -
columns
chr,pos,refandalt:chr,pos,ref,alt 12,6537866,C,T
The reference base can be omitted in any case (in the file you can leave the column out completely), but then BEscreen will not check if your reference base is the one it also finds. If these differ this indicates the usage of different reference genomes.
BEscreen will annotate the guides with several information. If you choose Use MANE select transcripts only, BEscreen will only use the MANE Select transcript for these annotations.
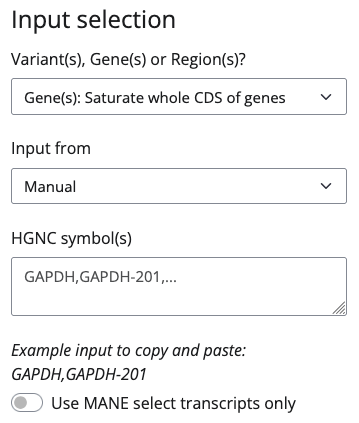
Gene(s)
If you don't want to start with pre-specified variants but rather want to design guides for a given gene or transcript, select Gene(s): Saturate whole CDS of genes. Next, select whether you want to input directly:
or from a CSV file:

Direct variant inputs need to be provided either as HGNC gene symbol (e.g., GAPDH) or transcript name (gene name followed by a hyphen and the transcript number; e.g., GAPDH-201) e.g. like this:
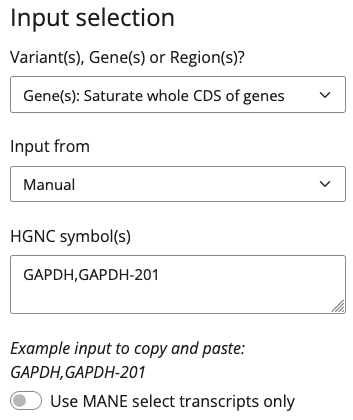
If you choose to input from file you can use the column symbol as shown in the following example:
- columns
symbol:symbol GAPDH GAPDH-201
Take note that the input is case sensitive, so enter "GAPDH" for the human gene, but "Gapdh" for the mouse gene.
If you use a gene name as input, BEscreen will use all transcripts for this gene to search and annotate the guides. If you choose Use MANE select transcripts only, BEscreen will only use the MANE Select transcript to search and annotate the guides. This results in guides filtered for the MANE transcript.
Negative and positive controls for your assay
The Gene(s) mode has the important functionality to design proper positive and negative controls for base editing experiments.
Positive controls
Base editors can introduce edits that are equivalent (or mostly equivalent) to a gene knockout - stopgain and splice-site edits. The earlier one of those edits is introduced in your CDS, the more likely this will have the same consequence as a knockout of the gene. You can filter for those guides using BEscreen's Filter options.
Negative controls
Even more important than positive controls are negative controls. BEscreen allows for two types of negative controls that are well-suited for base editing screens: non-editing guides (i.e., guides that bind but do not confer any edits) and guides that only introduce a synonymous mutation (i.e., a base edit that leads to a synonymous codon).
Region(s)
If you neither want to start with pre-specified variants nor given genes, you can use a complete genomic region. To this end, select Region(s): Set a genomic region to scan for editable bases. Next, select whether you want to input directly:

or from a CSV file:

If you want to input your region(s) directly, you need to use the form [CHROM]:[START]-[END] (e.g. 12:6536490-6537490 like this):

If you choose to input from file you can use the column region as shown in the following example:
- columns
region:region 12:6536490-6537490
Species
Next continue to the Species section and set your species by choosing a reference.

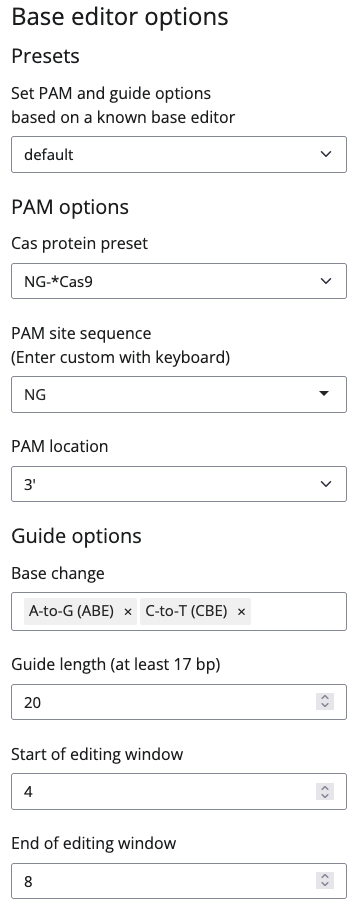
Base editor options
Presets
Select a Preset from a list of known base editors. This will set your PAM and guide options. You can alter any setting made be the presets to adjust BEscreen to your needs, if you experienced different behavior in your system. If you do so, the preset field will show (changed) to show you that you are not using the original preset settings anymore.
PAM options
Either use presets from different Cas protein presets to set your PAM site sequence and PAM location or define a custom PAM sequence using IUPAC code and choose, if it is located at the 5' or 3' end of the guide. If you do the latter, the preset field will show (changed) to show you that you are not using any Cas protein preset settings anymore.
Guide options
You can adjust the Base change, Guide length and the Start and End of editing window in Guide options. Be aware that the editing window is well characterized for some base editors, but not so for others. In general editing windows in general are not to be considered absolute - edits outside of the editing window might always happen. To account for this you could either increase the editing window or use our safety region option to analyze the bases adjacent to the editing window.
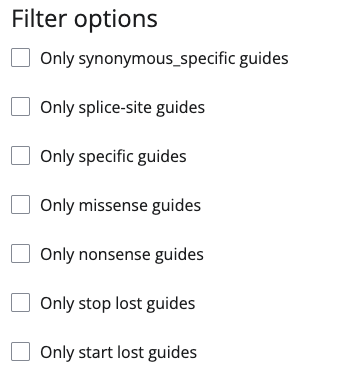
Filter options
If you are only interested in edits with specific consequences, you can set filters for synonymous_specific, splice-site, specific, missense, nonsense (stop gain), stop lost or start lost guides in the Filter options.
Additional annotations
BEscreen provides feature-rich output, but if you need more, you can add the annotation of Ensembl's Variant Effect Predictor and genome wide hits using NCBI's BLAST.
For VEP, you can select which consequences to show (BEscreen defaults to pick the top consequence according to VEP's order for the option --pick) and you can set the shown fields as known from the command line version of this tool. Experimentally, you can also forward other command lines flags to VEP, but be warned again: This is an experimental feature that in some cases might lead to unexpected results.
For BLAST you can choose only to BLAST the main chromosomes without contigs like e.g. GL000218.1 and KI270728.1.

Advanced options
Some more Advanced options are available. These differ for Variant(s), Gene(s) and Region(s). For all three you can set a safety region, if you know your base editor tends to edit outside its main editing window. BEscreen will not actively look for guides that edit in that region, but it will tell you, if the guides it found have an editable base in that area.
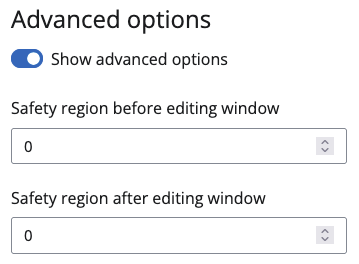
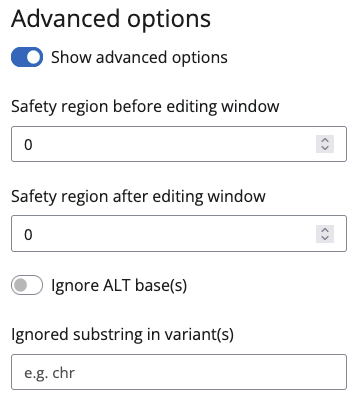
For Variant(s) and Gene(s) there are additional options:
Variant(s)
For Variant(s) you can set Ignore ALT base(s). If you for example provide an non-editable C to G mutation as input, BEscreen will not start searching for guides, but tell you that this edit is not possible. If you check Ignore ALT base(s), it will search for C to T mutations instead. This will most likely lead to different consequences as the original variant, but at least it lets you edit the position if guides for this position are found. You can also provide a string to be ignored in the Ignored substring in variant(s). If your variants are e.g. formatted with UCSC chromosome names (chr prefix) you can enter chr here to transform to Ensembl names. This only works for main chromosomes and it is discouraged to use, but is sometimes helpful to get a quick view on available guides.
Gene(s)
For Gene(s) you can select whether you want to actively search for splice-site guides or not. This is enabled per default.
Final decisions
For all Variant(s), Gene(s) and Region(s) modes BEscreen offers two different modes for the output table: collapsed or expanded. For Variant(s), "collapsed" will display one row per input variant (this can be more for some rsIDs that have ambiguous alternative bases), and "expanded" will display one row per guide (for some variants multiple guides are found). For Gene(s) and Region(s), "collapsed" displays one row per base editor guide combination, "and" expanded displays one row per variant that is introduced by a guide (guides can introduce one variant for every base in the editing window).

If you're happy with what you have done, click Design guides.

If not, click Reset all inputs and settings.

Download
Variant(s)
You can download your guides by clicking the corresponding download button.
For variants, you can download the full table or a table filtered for variants, for which guides were found.

Gene(s) and Region(s)
For the gene mode, you can download the full tables for editing guides or non-editing guides.

The output of BEscreen
The output of BEscreen is divided into one or two tables and embedded in igv.js showing the alignment of the found guides.
The table columns
Field separators
In general the different field separators for editing guides have different meanings:
Variant(s)
•: Separates the different guides that can introduce the intended edit. This applies to all columns except variant, base_change, symbol, strand and ref_match. This is the separator that gets expanded if you choose Expanded output.
;: Separates the effects of different edits introduced by the same guide. This applies to the columns consequence, codon_ref, aa_ref, aa_pos, aa_ref and aa_edit.
~: Separates transcripts and their properties having the same amino acid change resp. consequence. This applies to the columns aa_pos, transcript, exon_number, first_transcript_exon and last_transcript exon.
^: Separates transcripts and their properties having a different amino acid change resp. consequence. This applies to the columns synonymous_specific, consequence, codon_ref, aa_ref, aa_pos, aa_ref, aa_edit, splice_site_excluded, transcript, exon_number, first_transcript_exon and last_transcript exon.
Gene(s)
;: Separates the effects of different variants within the same guide. This applies to the columns variant, consequence, codon_ref, aa_ref, aa_pos, aa_ref and aa_edit. This is the separator that get expanded if you choose Expanded output.
~: Separates transcripts and their properties having the same amino acid change resp. consequence. This applies to the columns aa_pos, transcript, exon_number, first_transcript_exon and last_transcript exon.
^: Separates transcripts and their properties having a different amino acid change resp. consequence. This applies to the columns synonymous_specific, consequence, codon_ref, aa_ref, aa_pos, aa_ref, aa_edit, splice_site_excluded, transcript, exon_number, first_transcript_exon and last_transcript exon.
Region(s)
;: Separates different variants within the same guide. This applies to the column variant.
The columns
variant (not non-editing guides)
The variants used as input or found by BEscreen in the format [chromosome]_[genomic_position]_[reference_base]_[alternative_base]. The used variant will be added, if rsID or protein amino acid changes are used as input or if Ignore ALT base(s) has been selected.
base_change
The base editor class (ABE or CBE) or the generic base edit that can edit using this guide. In the variants mode one row for every variant<->base editor combination will be shown.
symbol (not for regions)
The gene affected by the edit.
guide, guide_with_pam
The guide sequence with and without the PAM sequence.
guide_chrom, guide_start, guide_end
The genomic location where this guide was found (it will bind that position on the reverse complement strand).
edit_window
The sequence of the editing window.
num_edits (not non-editing guides)
The number of edits in the editing.
specific (not non-editing guides)
If the guides only edits one or more bases in the editing window.
edit_window_plus (optional, if safety region selected)
The editing window with the safety region.
num_edits_plus (optional, if safety region selected; not non-editing guides)
The total edits in the editing window and safety region.
specific_plus (optional, if safety region selected; not non-editing guides)
If specific guides are also specific with the added safety region.
safety_region (optional, if safety region selected)
The safety region in capital letters with the main editing in minor letters.
num_edits_safety (optional, if safety region selected; not non-editing guides)
The number of additional edits in the safety region.
additional_in_safety (optional, if safety region selected; not non-editing guides)
If there are additional edits in the safety region.
ne_plus (optional, if safety region selected; only non-edition guides)
If non-editing guides are also non-editing with the safety region.
synonymous_specific (not non-editing guides; not for regions)
If the guide leads to an synonymous edit and is specific in the main editing window. BEscreen only annotates specific guides as synonymous_specific for safety reasons.
consequence (not non-editing guides; not for regions)
The consequence of the individual edits. Every edit is checked individually, even if they are in the same codon. Possible consequences are:
synonymous: The edit leads to the same amino acid.
missense: The edit leads to a different amino acid.
stopgain: The edit introduces an additional stop codon.
startlost: The edit deletes the start codon.
stoplost: The edit deletes the stop codon.
splice_site: The edit disrupts a splice site.
intron: The edit lies in a non-coding intronic region (mostly of target edits of guides introducing other edits in coding regions).
utr: The edit lies in a non-coding UTR region (mostly of target edits of guides introducing other edits in coding regions; sometimes BEscreen assumes a splice site, but then identifies that the edit is before the start or after the stop codon; such edits should be analyzed manually).
indefinite: BEscreen was not able to determine the codon (the user needs to analyze this edit manually).
complex: BEscreen was not able to determine the consequence (the user needs to analyze this edit manually).
not_in_cds: The edit lies outside of any CDS (only happens for variants).
strand
The strand where the guide binds (the reverse complement of where the guide sequence was found).
codon_ref, aa_ref (not non-editing guides; not for regions)
The affected codon(s) and amino acid(s) before the edit. Every edit is evaluated individually. No combined edits are taken into account yet.
aa_pos (not non-editing guides; not for regions)
The position of the affected amino acid in the translated protein of the affected transcript.
codon_edit, aa_edit (not non-editing guides; not for regions)
The affected codon(s) and amino acid(s) after the edit. Every edit is evaluated individually. No combined edits are taken into account yet.
splice_site_included (not non-editing guides; not for regions)
Whether the edit will possibly lead to splice site disruption.
originally_intended_ALT (optional, if you ignore ALT base(s); only for variants)
Whether the edit is the one you originally wanted.
ref_match (optional, if you ignore ALT base(s); only for variants)
Whether the input reference base is the same as BEscreen found in the genome. If not, this is a hint for a different genome assembly used in your analysis. BEscreen uses GRCh38.
edited_positions (not non-editing guides)
A string showing the positions of the edits in the guide sequence:
.: Just a base.
:: A base within the editing window.
number: An edit within the editing window with the value of the position of the edit.
,: A base within the safety region.
+: An edit within the safety region.
distance_to_center_variant (only for variants), distance_to_center (not non-editing guides)
The distance of the edits to the center of the editing window for the intended (distance_to_center_variant) and all (distance_to_center) edits.
transcript, exon_number, first_transcript_exon, last_transcript_exon (all not for regions)
The transcript name and exon number affected by the edit. Also, the first and last exons of the affected transcript are shown. Transcripts with the same amino acid change will be combined by ~. Transcripts with a different same amino acid change will be separated by ^.
blastcount (only, if BLAST selected)
The number of perfect genomic alignments for this guide identified using NCBI's BLAST+ algorithm.
VEP_* (not non-editing guides; only, if VEP selected)
Additional annotations according to Ensembl's Variant Effect Predictor (VEP). The columns can be customized. All columns produced by VEP have a VEP_ prefix. As a default, only the consequence with the highest priority according to Ensembl is shown for enhanced readability. This can be changed to show all consequences (separated by ,), the most severe or a summary for the given variant. For the variants mode, only the variant used as input is shown. For genes mode, all variants introduced by a guide are shown. Consequences for different variants are separated by ; in the genes mode.
Example view of the tables
Here is an example how the output table for editing and non-editing guides look for the genes GAPDH with default settings (you can enlarge any of those tables by clicking the < > symbol in the right lower corner):
The editing guides:
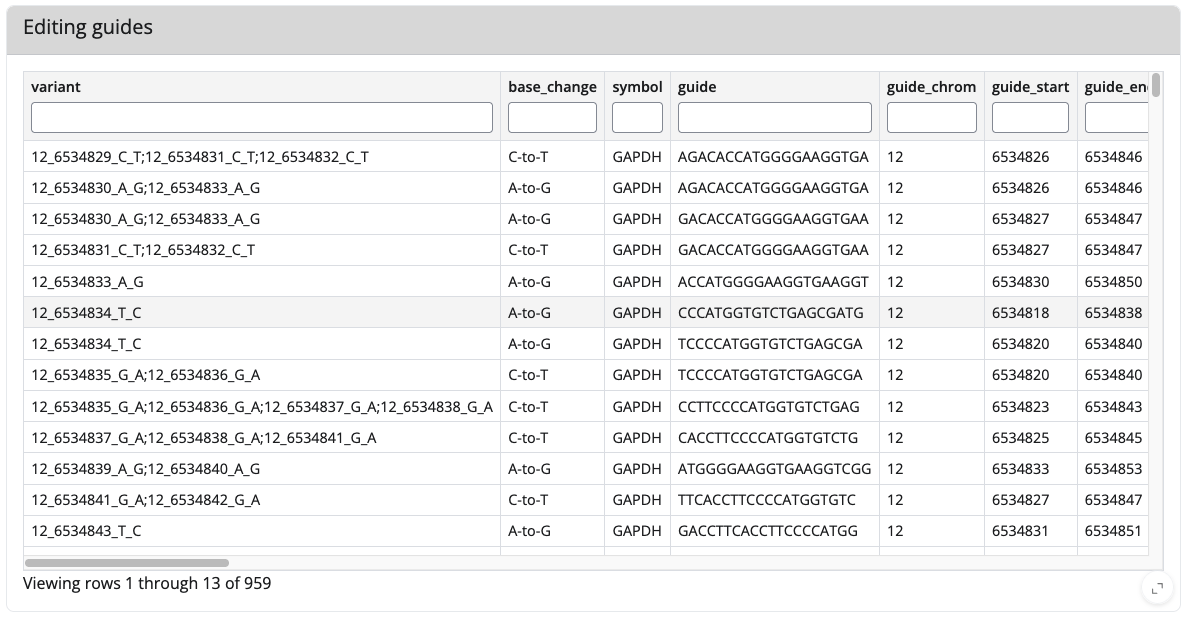
The non-editing guides:

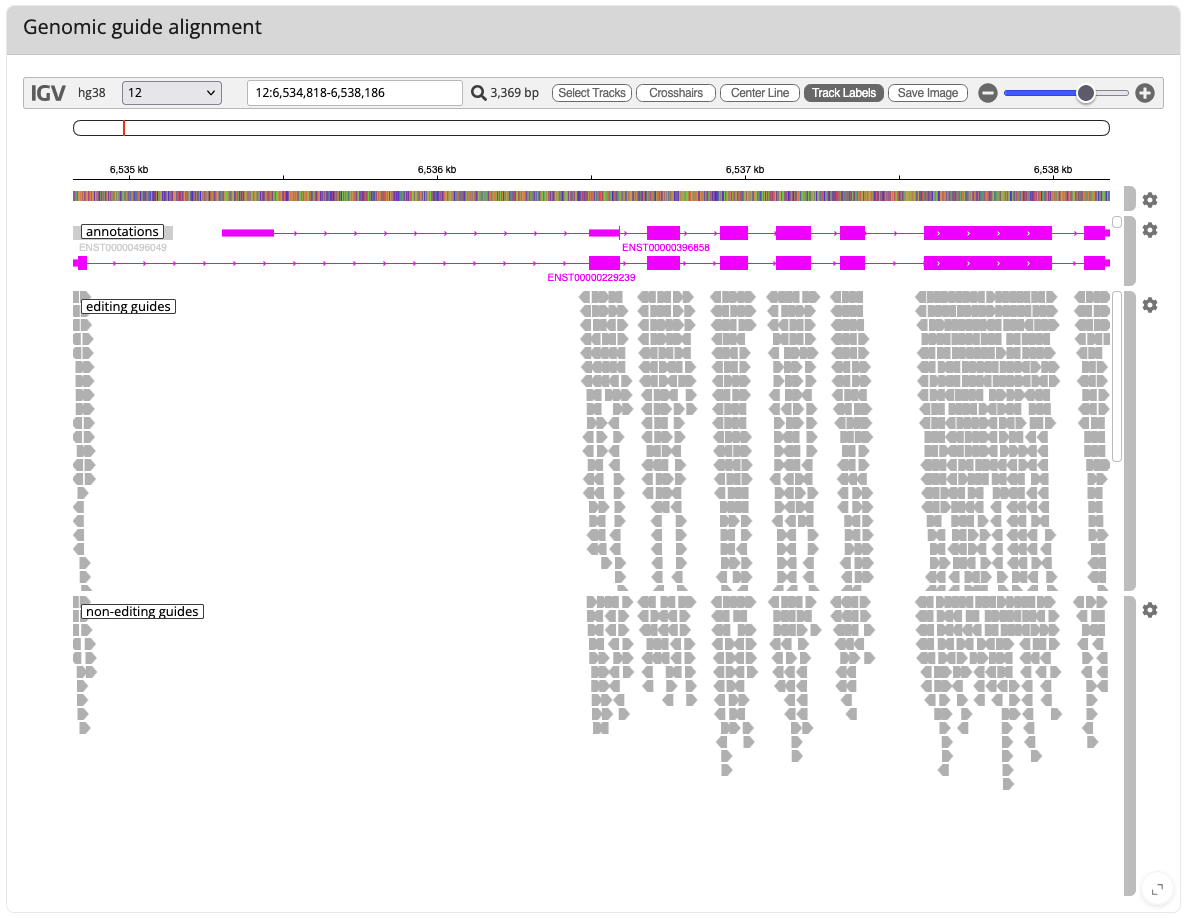
Example view of igv.js
And here is an exmaple view of igv.js showing you where your guides bind in the genome (here again for GAPDH; you can also enlarge igv.js by clicking the < > symbol in the right lower corner).
How to use BEscreen
Here, we give you a little introduction, how you could use BEscreen to design guides for the APOE gene as already outlined in the original BEscreen publication.
Design guides for known and unknown APOE variants
Three highly prevalent variants of APOE exist, termed APOE2, APOE3, and APOE4, which are defined by two SNPs. The first SNP (rs429358) is a T to C mutation on chromosome 19 at position 44908684 leading to a missense mutation of the codon TGC (coding for cysteine) to CGC (coding for arginine), resulting in a APOE-C130R mutation. The other SNP (rs7412) corresponds to a C to T substitution at position 44908822 leading to a APOE-R176C mutation.
Designing guides for these variants using BEscreen's variants mode
You can use BEscreen’s Variant(s) mode to identify guides that mediate the introduction of the above mentioned mutations. To do so, just keep all the settings at default level. This will use the Variant(s) mode and search for guides for A-to-G (ABE) and C-to-T (CBE) edits for an NG PAM, with a guide length of 20 and an editing window in the positions 4 to 8. These are the settings we use in our lab for our base editors. If those don't suit your base editor(s) adjust them accordingly. This you should check in the literature (or known from your experience).
Then simply use the two rsIDs as input for Variant(s) and hit Design guides at the bottom of the page. BEscreen identifies one guide suitable for the rs429358 edit and two guides for the s7412 edit (this might of course be different, if you changed the base editor, PAM or guide options).
BEscreen's tabular output shows that this guide introduces a second edit changing the upstream GTG codon to GCG, resulting in an additional missense mutation (V129A) additionally to the desired C130R. You can identify the wanted edit by the capitalized consequence, codon_ref, aa_ref, codon_edit and aa_edit.
For the rs7412 SNP, BEscreen identified two suitable guides mediating the desired R176C mutation.
By entering variants at the protein instead of nucleotide level (APOE-C130R instead of rs429358 and APOE-R176C instead of rs7412), BEscreen searches for whether edits in any of the three bases making up the affected codon achieve the intended protein consequences by editing a different base. This is not the case for these variants with our settings.
The identified guides you can now use for your experiments, but only after you also have some proper positive and negative controls. Those can be designed using BEscreen's Gene(s) mode, which the next chapter will cover.
Designing guides for these variants using BEscreen's variants mode
To design an sgRNA library introducing these and all other possible edits into APOE, you can use BEscreen’s Gene(s) mode. Just switch to the Gene(s) panel with the Variant(s), Gene(s) or Region(s)? selector right at the top in the Input selection. Enter APOE as input for HGNC symbol(s), keep all other settings as they are (or change them, if you need to) and again hit Design guides at the bottom of the page.
BEscreen just designed 1,065 NG PAM-dependent editing guides. Using the different filters BEscreen identifies 506 guides which mediate only one edit and are thus annotated as specific, 799 guides mediating at least one missense mutations, 131 guides mediating only one specific synonymous edit (synonymous_specific), 20 splice site editing, 90 nonsense, 9 start lost, and 3 stop lost introducing guides.
Positive and negative controls
In addition to these editing guides the Gene(s) mode designed 402 binding but non-editing guides. These non-editing guides and guides mediating specific synonymous edits are valuable as negative controls in a base editing screen, while guides leading to splice site disruptions or nonsense (stop gain) mutations can serve as positive controls. These controls you can (and probably should) of course also use for the guides identified in BEscreen's Variant(s) mode. Thus, BEscreen just designed you a complete library for evaluating known and unknown variants in the APOE gene with minimal and convenient input requirements.
Just download the tables or pick the sequences directly and start cloning your guide(s) and/or library!
Is BEscreen available as local command-line version?
Yes, you can find a command-line version that you can run locally at github.com/ostendorflab/bescreen.
Can you add feature XYZ?
We'll be happy to take a look! Reach out to us by mail (bescreen@ostendorflab.org) and we'll get back to you. You can also open an issue in the Github repository.
How do I cite BEscreen?
BEscreen: a versatile toolkit to design base editing libraries has been published in the Nucleic Acids Research Web Server Issue 2025. Please cite this article, if you are using BEscreen:
Philipp G Schneider, Shuang Liu, Lars Bullinger, Benjamin N Ostendorf, BEscreen: a versatile toolkit to design base editing libraries, Nucleic Acids Research, 2025;, gkaf406, https://doi.org/10.1093/nar/gkaf406
Can I use BEscreen for my work?
BEscreen itself is licensed under CC BY 4.0![]()
![]() . The third party tools Ensembl Variant Effect Predictor (VEP), NCBI BLAST and igv.js (used for visual guide representation) have their own licenses. If you use these, please check if their licenses are compatible with your work.
. The third party tools Ensembl Variant Effect Predictor (VEP), NCBI BLAST and igv.js (used for visual guide representation) have their own licenses. If you use these, please check if their licenses are compatible with your work.
You can reach us by email. For more information about our lab, take a look here. For feature requests or bug reports please either email or create an issue in the Github repository.
Changelog
2026-03-18: Included the possibility to search for variants for intron-spanning codons in the variants mode.
2026-03-18: Fixed a bug leading to an exception where aa mutations cannot be introduced with a single base change in the variants mode.
2026-03-17: Fixed a bug for gene names containing hyphen in the variants mode.
2026-03-17: Fixed a bug which lead to an infinite loop when giving amino acids with intron-spanning codons in the variants mode. Right now these amino acids cannot be used as input, but at least they are recognized as intron-spanning now.
2026-02-10: Added Ensembl release 98 for human GRCh38.
2025-12-15: Fixed a bug after using 5' PAMs. This bug led to wrong results after using 5' PAMs once even for 3' PAMs.
2025-12-14: Changed behavior of VEP annotations for variants mode: Now showing information for all variants in variant_and_bystanders.
2025-12-14: Added new column variant_and_bystanders for variants mode showing all edits of a guide in the variant format.
2025-11-25: Fixed a bug for gene names containing hyphen in the genes mode.
2025-10-14: Fixed a bug for the consequences in the variants mode, if no CDS was found.